SpliceGrapher User’s Guide¶
Introduction¶
Overview¶
SpliceGrapher predicts alternative splicing patterns and produces splice graphs that capture in a single structure the ways a gene’s exons may be assembled. It enhances gene model annotations using evidence from next-generation sequencing and EST alignments. With SpliceGrapherXT (version 0.2.3), we introduced the ability to convert splice graph predictions into transcript predictions using IsoLasso or PSGInfer.
Downloading and Installation¶
- You may download SpliceGrapher from http://sourceforge.net/projects/splicegrapher
- Requires PyML version 0.7.9 or higher for users who wish to classify splice sites.
- Requires matplotlib version 1.1.0 or higher for users who wish to use the extensive graphics tools.
- Requires pysam version 0.5 or higher for users who wish to work with both BAM and SAM formats.
- The optional IsoLasso pipeline requires IsoLasso version 2.6.1 or higher, along with the gtfToGenePred and genePredToBed programs from UCSC
- The optional PSGInfer pipeline requires PSGInfer version 1.1.3
Currently Unix/Linux and Mac OS-X are supported. A setup.py script is provided so installation is standard for python:
python setup.py build
python setup.py install
To check that SpliceGrapher is installed correctly, run the python interpreter and type
>>> import SpliceGrapher
>>> SpliceGrapher.__version__
'0.2.7'
To test your installation more thoroughly, use the test script in the SpliceGrapher examples sub-directory:
cd examples
run_tests.sh
Tutorial¶
Before getting into SpliceGrapher’s nuts and bolts, we begin with a few examples
to demonstrate its key features. In the directory where you installed SpliceGrapher
you will find a sub-directory called tutorial that contains all the data you will
need for the following examples. Be sure that SpliceGrapher’s scripts are in your path.
Note that all of the examples use the SpliceGrapher.cfg file in the tutorial
directory. This contains the paths to the default reference genome (a_thaliana.fa.gz)
and the default gene models (a_thaliana.gff3.gz). See The SpliceGrapher Environment for details.
Example 1: Create Classifiers
In this example you will create accurate models for canonical (GT-AG) and semi-canonical
(GC-AG) splice sites for the plant Arabidopsis thaliana. To reduce classifier training
time you may also want to set the number of examples to something relatively small such
as -n 400 (the default is 2000). Create the classifiers by running the
build_classifiers.py script as follows:
build_classifiers.py -d gt,gc -n 400
The program shows you the SpliceGrapher scripts it uses for each step in the process. Below is output from a typical run:
Creating SVM models for splice sites
09:45:10 generate_splice_site_data.py -d gt -n 200 -o gt_don_training.fa -r splice_site_report.txt
09:45:20 generate_splice_site_data.py -d gt -n 200 -o gt_don_neg.fa -N
09:45:29 cat gt_don_neg.fa >> gt_don_training.fa
09:45:29 rm gt_don_neg.fa
09:45:51 generate_splice_site_data.py -d gc -n 200 -o gc_don_training.fa
09:46:01 generate_splice_site_data.py -d gc -n 200 -o gc_don_neg.fa -N
09:46:10 cat gc_don_neg.fa >> gc_don_training.fa
09:46:10 rm gc_don_neg.fa
09:46:31 generate_splice_site_data.py -a -d ag -n 200 -o ag_acc_training.fa
09:46:40 generate_splice_site_data.py -a -d ag -n 200 -o ag_acc_neg.fa -N
09:46:49 cat ag_acc_neg.fa >> ag_acc_training.fa
09:46:49 rm ag_acc_neg.fa
09:47:12 zip classifiers.zip ??_???.cfg ??_???.svm ??_???.fa
09:47:12 rm ??_tmp_e*_i*.fa
Finished.
When the script finishes, you may check classifier performance by looking for the ROC scores in their corresponding .cfg files:
grep roc *.cfg
Scores typically will be above 0.90:
ag_acc.cfg:roc_score = 0.935625
gc_don.cfg:roc_score = 0.94925
gt_don.cfg:roc_score = 0.969875
The script packages these classifiers into the classifiers.zip file in the local directory.
Example 2: Filter Alignments
For this example you may use the classifiers you created in Example 1, or
you may simply copy the Arabidopsis_thaliana.zip file
from classifiers sub-directory of your SpliceGrapher distribution.
This example uses the following files:
- Alignment output (SAM format):
alignments.sam.gz - A. thaliana classifiers (ZIP format):
classifiers.zip(from Example 1 above) orArabidopsis_thaliana.zip
To filter the alignment output, run the sam_filter.py script:
sam_filter.py alignments.sam.gz classifiers.zip -o filtered.sam
For this example we included several false-positive alignments to illustrate the filtering process.
If you run sam_filter.py with verbose turned on (-v option) you will see that these
have been filtered out of the output file.
Example 3: Predict a Splice Graph
This example demonstrates SpliceGrapher’s prediction modules. In this case we will use it to predict a graph for the gene AT2G04700 that we selected as an illustrative example. This example uses the following files:
- Alignment output (SAM format):
filtered.sam - Plotter configuration file:
AT2G04700_plot.cfg
To predict a graph for this gene, run the predict_splicegraph.py script:
predict_splicegraph.py AT2G04700 -d filtered.sam -o AT2G04700.gff
Predicting a graph for a single gene is useful, but in most cases you will want to make predictions for a collection of genes (or genome-wide). In that case you will want to use the predict_graphs.py script that is part of the extended example in section RNA-Seq Analysis Example.
There are two ways to view the resulting graph: view_splicegraphs.py and plotter.py.
The first script allows you to view a splice graph on your screen or, if you prefer, save it
to a file. plotter.py is designed for file output and provides a more extensive set of
options. We have provided an example plotter configuration file to get you started:
view_splicegraphs.py AT2G04700.gff -L
plotter.py AT2G04700_plot.cfg
RNA-Seq Analysis Example¶
On the SpliceGrapher SourceForge website we have included RNA-Seq-tutorial.tgz that contains files so that you can perform a complete RNA-Seq analysis to get better acquainted with the software. In this tutorial you will filter alignments, generate predictions from the alignments, generate plots for a couple of genes and compare statistics for the original gene models with the predicted graphs. We recommend that you create a separate directory to run this example to make it easier to keep track of what you’re doing.
Download and unpack the tar file¶
First download the tutorial file from SourceForge into the directory you are using to run the example and unpack the tarball as follows:
tar xfz RNA-Seq-tutorial.tgz
You should find the following files for the model plant A. thaliana:
accepted_hits.sam
Arabidopsis_thaliana.zip
a_thaliana.fa
a_thaliana_reduced.gtf
a_thaliana_reference.gtf
AT5G18280.cfg
The accepted_hits.sam file contains alignments we produced by running Tophat on a set of simulated reads. Arabidopsis_thaliana.zip contains the files for splice-site classifiers used in the filtering step. The file a_thaliana.fa containes the reference genome in FASTA format. There are two files containing gene models: a_thaliana_reduced.gtf contains a single representative transcript for 1000 randomly-sampled genes from A. thaliana, while a_thaliana_reference.gtf contains the complete TAIR10 gene models for these same genes. Finally, AT5G18280.cfg is a configuration file to use with the plotter.py script once splice graphs have been created.
Filter Tophat alignments¶
Given a set of alignments in accepted_hits.sam, the first step is to filter the alignments to eliminate possibly spurious spliced alignments. The sam_filter.py script is used to perform this task:
sam_filter.py accepted_hits.sam Arabidopsis_thaliana.zip -f a_thaliana.fa
-m a_thaliana_reference.gtf -v -o filtered.sam
The output should look similar to the following:
Loading FASTA records from a_thaliana.fa
Found 7 FASTA records in a_thaliana.fa
loaded classifier for 'ag' acceptor sites
loaded classifier for 'gc' donor sites
loaded classifier for 'gt' donor sites
Loading and validating gene models from a_thaliana_reference.gtf
..
Validating junctions in accepted_hits.sam
....|....|....|....|....|....|....|....
Found 554,708 ungapped and 271,298 spliced alignments in accepted_hits.sam
7,654 (99.4%) TP and 45 (0.6%) FP junction alignments out of 7,699 total
40 (47.1%) TP and 45 (52.9%) FP novel junction alignments out of 85 total
Also found 78 intergenic and 40 transgenic junctions
Predict splice graphs¶
Next predict splice graphs using the reduced gene models and the filtered alignments created in the previous step. When predicting graphs for multiple genes, use the predict_graphs.py script as follows:
predict_graphs.py filtered.sam -m a_thaliana_reduced.gtf -v -d predicted
The reduced gene models in a_thaliana_reduced.gtf consist of a single representative transcript per gene, while the SAM alignments represent a much broader set of transcripts from the complete gene models. Later we will compare the predicted graphs with graphs generated from the complete gene models in a_thaliana_reference.gtf. Output from the predict_graphs.py script should appear as follows:
Loading and validating gene models from a_thaliana_reduced.gtf
Initializing SAM input from filtered.sam
Found 5 chromosomes in common between a_thaliana_reduced.gtf and filtered.sam:
1, 2, 3, 4, 5
Generating predictions:
starting chromosome 1
loading alignment records from filtered.sam
predicting splice graphs .
starting chromosome 2
loading alignment records from filtered.sam
predicting splice graphs
starting chromosome 3
loading alignment records from filtered.sam
predicting splice graphs
starting chromosome 4
loading alignment records from filtered.sam
predicting splice graphs
starting chromosome 5
loading alignment records from filtered.sam
predicting splice graphs .
Finished: splice graphs were modified for 847 / 1,000 genes.
Generate plots¶
Now you can experiment with the plotting utility plotter.py using the plotter configuration file AT5G18280.cfg provided in the tar file. Use the -v option to monitor its progress:
plotter.py AT5G18280.cfg -v
Notice that this command takes some time to run. A significant part of the time is taken reading the SAM input file. To speed this process, you can split the SAM file into smaller pieces, one file per chromosome:
sam_split.py filtered.sam
This should create the files 1.sam, 2.sam, 3.sam, 4.sam and 5.sam. To improve plotting speed, modify the AT5G18280.cfg file and change the name of the SAM file it uses. There are two locations in the file where you will want to change filtered.sam to 5.sam. Both lines appear as follows:
source_file = 5.sam
The plotter script produces an output file AT5G18280.pdf. If you look at the PDF file you will notice that the figure includes the splice graph for the reduced gene model, the spliced alignments and the read coverage. At this point it is instructive to add the predicted graph to the plot, which you can do by adding the following section to the configuration file (assuming you placed your predicted graphs in the predicted directory):
[MyPrediction]
plot_type = splice_graph
source_file = predicted/5/AT5G18280.gff
title_string = AT5G18280 Prediction
To make the splice graphs adjacent in the final figure, insert this section right before the [SpliceJunctions] section in the configuration file. Now the plot will show the splice graph for the reduced gene model as well as the predicted splice graph, allowing you to see the exons added by the prediction software.
Depths files¶
We can now repeat the exercises in the last two sections using SpliceGrapher depths files in place of SAM files. First convert the filtered SAM alignments into a depths file:
sam_to_depths.py filtered.sam -o filtered.depths
If you do a directory listing you will see that the depths file is much smaller than the SAM file, meaning that predictions and plotting should take less time. Now you can use the depths file in place of the SAM file to predict splice graphs:
predict_graphs.py filtered.depths -m a_thaliana_reduced.gtf -v -d depths_predicted
You may also wish to rerun plotter.py by first modifying the configuration file and
replacing all references to SAM files with filtered.depths.
Compare predictions with complete gene models¶
We can also compare the predicted splice graph with the complete gene models. The SAM alignments were simulated from the complete gene models, so we would like to know how closely the predicted graphs match the complete gene models. The full gene models for these genes are in the fila a_thaliana_reference.gtf. To add these to our plot, we first need to generate splice graphs from the gene models using the gene_model_to_splicegraph.py script:
gene_model_to_splicegraph.py -m a_thaliana_reference.gtf -a -S -d reference
This will create splice graphs in the same kind of directory structure as for a set of predictions. We can now add the splice graph for the complete gene model to the plot configuration file by adding a new section:
[CorrectGraph]
plot_type = splice_graph
source_file = reference/5/AT5G18280.gff
title_string = AT5G18280 Gene Model
As before, to keep the splice graphs adjacent in the final figure, insert this section right before the [SpliceJunctions] section in the configuration file. Now when you generate a plot, you can see the representative transcript, the predicted graph and the complete gene model all on the same figure.

Generate statistics¶
In addition to viewing individual genes, it is also worthwhile to generate statistics from a set of splice graphs using the splicegraph_statistics.py script. This script accepts a list of splice graphs as input, that we can create easily using the Linux find command:
find predicted -name "*.gff" > predicted.lis
find reference -name "*.gff" > reference.lis
Once we have created these lists, we can generate statistics for the two sets of graphs. The output from these commands should be similar to the following for the predicted graphs:
splicegraph_statistics.py predicted.lis
Alternative Splicing Statistics for 1,000 Graphs (780 with AS)
Intron Retention Skipped Exon Alt. 5' Alt. 3' Total
345 (28.7%) 180 (15.0%) 257 (21.4%) 420 (34.9%) 1,202
For the reference gene models, the statistics are different:
splicegraph_statistics.py reference.lis
Alternative Splicing Statistics for 1,000 Graphs (863 with AS)
Intron Retention Skipped Exon Alt. 5' Alt. 3' Total
443 (29.7%) 184 (12.3%) 327 (21.9%) 536 (36.0%) 1,490
Starting with version 0.2.7, it is also possible to generate a table of statistics for all genes, as follows:
genewise_statistics.py predicted.lis > genewise_AS.tab
genewise_statistics.py reference.lis > reference_AS.tab
The output format makes it easy to compare samples on a gene by gene basis:
Gene Alt5 Alt3 ES IR Total
AT1G01020 0 0 0 1 1
AT1G01040 1 0 0 0 1
AT1G01060 0 2 1 1 4
AT1G01070 0 0 0 1 1
Note: the statistics for SpliceGrapherXT’s predicted graphs are slightly lower than for the complete gene models. This is because predictions were based on a single representative transcript for each gene (in other words, no AS events to start with), and SAM alignments that were simulated from the complete gene models. Thus the predictions reflect only those known AS events that were present in the simulated alignments.
Generate plots for other genes¶
Earlier we developed a configuration file for the gene AT5G18280. Assuming this configuration has the characteristics we want, it is relatively simple to use the same configuration for another gene. First, copy the configuration file to create a new one:
cp AT5G18280.cfg AT2G45960.cfg
Next, replace every instance of AT5G18280 with AT2G45960 in the new file. You will also need to replace every reference to the chromosome subdirectory /5/ with /2/:
predicted/2/AT2G45960.gff
reference/2/AT2G45960.gff
Finally, replace every instance of 5.sam with 2.sam:
source_file = 2.sam
Now you can generate a new figure for the gene:
plotter.py AT2G45960.cfg

The SpliceGrapher Environment¶
Throughout the this guide we describe a variety of tasks that require different input files. By far the most common files are a reference sequence (a FASTA file) and a gene model (either GFF3 or GTF format).
Environment Variables¶
You may specify default paths for these files by setting
the environment variables SG_FASTA_REF and SG_GENE_MODEL. For example, in C shell
this might look like:
setenv SG_FASTA_REF "/home/o_sativa/sequences/o_sativa.fasta"
setenv SG_GENE_MODEL "/home/o_sativa/annotations/o_sativa.gff3"
SpliceGrapher will use these default paths when they are not provided to a script on the command line or in a configuration file.
SpliceGrapher Configuration File¶
SpliceGrapher programs and modules also use the configuration file SpliceGrapher.cfg
to locate the genomic reference and gene model files.
Below is an example of a configuration file.
[SpliceGrapher]
GENE_MODEL = /home/o_sativa/annotations/o_sativa.gff3
FASTA_REFERENCE = /home/o_sativa/sequences/o_sativa.fasta
SpliceGrapher scripts look for SpliceGrapher.cfg in your PATH.
When working with data for different organisms, it is often convenient to
place a SpliceGrapher.cfg file for each organism in its own directory.
Then one can change SpliceGrapher’s defaults simply by changing directories.
SpliceGrapher scripts will first use any paths specified on the command line,
followed by those found in a configuration file
and then those given as environment variables.
Splice Site Classifiers¶
For RNA-Seq data we are concerned with two kinds of alignments: ungapped alignments and splice junction alignments. An ungapped alignment is one in which a read aligns completely within an exonic region in the reference sequence. Most short-read alignment algorithms can perform exonic alignments. A splice junction alignment is one in which part of a read aligns to the 3’ end of one exon and the remainder aligns to the 5’ end of another exon, usually within the same gene. However, many spliced alignment programs use only rudimentary heuristics to identify correct splice sites. SpliceGrapher’s approach is to filter these junctions using highly accurate splice-site classifiers.
SpliceGrapher comes with over 100 pre-built
classifiers for a variety of different species. These are provided as
.zip files located in the classifiers directory. You may also build
your own classifiers using the build_classifiers.py script.
Building Classifiers¶
To simplify the process of constructing classifiers and to provide an example script
you may use to create your own pipeline, SpliceGrapher includes the script
build_classifiers.py. Assuming you have established paths for a FASTA
reference sequence a gene model file SpliceGrapher.cfg file, you can generate
a complete set of classifiers for filtering spliced alignments with one command:
build_classifiers.py
The script performs the following steps:
- Generates training data for each splice site dimer.
- Selects optimal parameters for each classifier.
- Stores the data for each classifier in a
.zipfile
The script accepts the following options:
| Option | Value | Description |
|---|---|---|
| –commands | Show but don’t run commands | |
| -a | dimer list | Acceptor site dimers to predict |
| -d | dimer list | Donor site dimers to predict |
| -f | file path | Fasta reference file |
| -m | file path | GFF3 gene model annotation file |
| -n | examples | Total number of examples to use for training |
| -l | file path | Optional output log file |
By default the script builds classifiers only for canonical GT donor sites and AG acceptor sites. To build a classifier for semi-canonical GC donor sites as well, simply provide a list to the script:
build_classifiers.py -d gt,gc
In each step of this pipeline, the main script calls other SpliceGrapher
scripts to generate data and train classifiers.
By using the --commands option, you can see the commands that would be generated
without actually running them. This is instructive when learning how to use each
of the scripts individually. Each of the scripts used in build_classifiers.py
is described in the following sections.
(For more information on how these classifiers are constructed, please see [BENHUR2008] and http://pyml.sourceforge.net/).
For the splice site prediction modules SpliceGrapher uses support vector machines (SVM) implemented in the PyML package. To create a set of splice-site classifiers, we suggest taking the following steps:
- Identify most common splice-site dimers for the species
- Generate positive, negative training data sets
- Find optimal classifier parameters for each splice site
Each of these procedures is described in the following sections.
Identifying Splice-Site Dimers¶
The workhorse script generate_splice_site_data.py can identify frequently-occurring
splice-site dimers and generate positive and negative example
sequences for any given dimer. The script can produce a report of all splice
site dimers and splice junctions found in a set of gene models. The most
frequently occurring splice site dimers are then candidates for constructing
classifiers for predicting novel splice sites in a genome. The command is simply:
generate_splice_site_data.py -r
Below is an example of a report, truncated for readability:
Breakdown of donor sites for 122535 introns:
GT: 121165 (98.88%)
GC: 1248 ( 1.02%)
AT: 84 ( 0.07%)
GA: 13 ( 0.01%)
TT: 8 ( 0.01%)
Breakdown of acceptor sites for 122535 introns:
AG: 122419 (99.91%)
AC: 76 ( 0.06%)
AT: 9 ( 0.01%)
Breakdown of splice junctions:
GT-AG: 121132 (98.8550%)
GC-AG: 1248 ( 1.0185%)
AT-AC: 73 ( 0.0596%)
GA-AG: 13 ( 0.0106%)
TT-AG: 8 ( 0.0065%)
Given a list of splice-site dimers and their frequencies, one can then choose an optimal set that balances the potential number of successful predictions with the overhead of training a classifier for each dimer.
Generating Training Data¶
The generate_splice_site_data.py script is also used to generate positive
and negative examples for training SVMs. Positive examples are simply examples
of the given dimer found at splice sites in the gene model. To generate negative
examples SpliceGrapher uses the procedure outlined in [RATSCH2005]: it
looks for all examples of the given dimer within each gene and uses as negative
examples those that don’t appear in splice sites.
| [RATSCH2005] | Accurate splice site detection for Caenorhabditis elegans. G. Rätsch and S. Sonnenberg, Oxford Bioinformatics, vol. 21, pp i369-i377, 2005. |
To generate positive examples for a given donor-site dimer such as GT, use
the following command:
generate_splice_site_data.py -o gt_positive.fa -d GT
To generate examples for acceptor sites, use the -a option:
generate_splice_site_data.py -o ag_positive.fa -d AG -a
As with other scripts, the default gene model and FASTA reference are provided
in SpliceGrapher.cfg variables, and output is written to stdout by
default.
To generate negative examples, simply add the -N option to the command:
generate_splice_site_data.py -o gt_negative.fa -d GT -N
By default, output splice site sequences will include 100nt on either side of a
splice-site dimer, but will not include the dimer itself. To include splice-site
dimers in output sequences, use the -D option. To change the window size
(for example, 30nt on either side), use the -W option:
generate_splice_site_data.py -o gt_positive.fa -d GT -W 30
Note that the window size will reduce the number of possible SVM parameter
combinations (described in Optimizing Classifiers) but may also reduce SVM
performance if the windows are too small. Positive examples for any dimer tend
to be relatively rare, while negative examples are plentiful. In fact, there may
orders of magnitude more negative examples than positive, which would be
overkill for training a good classifier. To set a limit on the number of
negative examples generated, use the -n option. When a limit is set, the
script will perform the following random selection procedure a fixed number of
times.
First it selects a gene at random from the genome. Once a gene has been
selected, it samples positions at random from within the gene until it finds the
appropriate dimer at a position other than a known splice site. Once the
appropriate number of negative examples have been randomly selected, the script
halts. Thus to generate a sample of 10,000 negative GT examples, use the
command:
generate_splice_site_data.py -o gt_negative.fa -d GT -N -n 10000
Optimizing Classifiers¶
SpliceGrapher uses support-vector machines (SVMs) to classify splice sites. More specifically it uses SVMs with a weighted-degree kernel that has proved especially successful at discriminating real splice sites from spurious ones. While these models are well-suited for predicting novel splice sites, there are a number of parameters that that can influence SVM performance for a specific kind of splice site. Briefly, these are:
- intronic and exonic sequence length on either side of a site
- range of k-mer sizes to use
- size of shifts to allow when assessing k-mers
- number of mismatches to allow
- SVM regularization parameter, C
For an overview and tutorial on SVMs with weighted-degree kernels, see [BENHUR2008].
| Option | Value | Description |
|---|---|---|
| -a | Interpret splice sites as acceptors | |
| -e | EXONSIZE | List of exon sizes to try (e.g., ‘20,25,30’) |
| -C | CLIST | List of regularization constants for SVM (e.g., ‘0.1,1.0,10.0’) |
| -i | INTRONSIZE | List of intron sizes to try (e.g., ‘20,25,30’) |
| -l | LOGFILE | Optional log file for tracking performance |
| -k | KFOLDS | Number of folds to run cross-validation |
| -m | MINK | Set of minimum k values for kernel (e.g, ‘1,2,3’) |
| -M | MAXK | Set of maximum k values for kernel (e.g., ‘3,4,5’) |
| -N | Turn normalization OFF | |
| -p | Apply a mismatch profile | |
| -P | PREFIX | Optional prefix for output files |
| -S | SHIFT | List of maximum shift values |
To simplify this process, SpliceGrapher includes the script select_model_parameters.py.
This script trains a weighted-degree kernel SVM on
labeled FASTA sequences generated by generate_splice_site_data.py and runs
cross-validation for a variety of parameter settings. As the program proceeds,
it saves the best model and parameter configuration. The basic format of this
command is
select_model_parameters.py FASTA-training-data dimer
For example:
select_model_parameters.py gt_training.fa gt
This runs cross-validation on a model with just the default parameters
(exon lengths of 8nt, 12nt and 16nt;
intron lengths of 15nt, 20nt and 25nt;
minimum k-mer size 1;
maximum k-mer 1, 2 or 3;
no mismatches;
shift sizes 0 and 1, and
C = {0.01,0.1,1,10,100}).
By default, cosine normalization is applied to kernel
vectors, but this may be turned off using the -N flag.
The script accepts a list of values to try for every SVM parameter. For example,
to try several shift values, one might use '-S 0,1,2,3'. Note, however, that
the number of parameter combinations grows geometrically with each set of
values. Since it can take a long time to run cross-validation on an elaborate
model, the program accepts a maximum of 100 parameter combinations per run.
Intron and Exon Sequence Lengths¶
When generate_splice_site_data.py generates training data, it uses the same
window size on either side of a splice site. This permits the model selection
program to try different combinations of intron and exon sequence lengths up to
this window size.
Note: in theory, one could use windows that are hundreds or thousands of nucleotides long. But as window sizes increase, the time required for cross-validation and for classification can increase dramatically. Experience has shown that classifier performance often does not improve significantly beyond a range of 30-40nt on either side of a splice site. The parameter selection script imposes a hard limit of 50nt on either side of a junction. If you wish to experiment with longer sequences you will need to change the hard-coded limit.
To identify the best intron and exon sequence lengths, run the model
selection algorithm with a series of possible lengths for each. For example, if
one wishes to find the best model performance for a donor GT splice site
with exon sequence sizes from 10 to 30 and intron sequence sizes from 10 to 40,
the command would then be:
select_model_parameters.py gt_training.fa gt -e 10,20,30 -i 10,20,30,40
To experiment with parameters, simply include in the same command values for those parameters you wish to try:
select_model_parameters.py gt_training.fa gt -e 10,20,30 -i 10,20,30,40 -m 1,2 -M 2,3,4
Note: despite one’s best efforts, some parameter settings may cause an SVM to fail to converge. Often this may be corrected by adding training data or by using different parameter values. For this reason we recommend working with only a few parameters at a time, taking care to save the resulting output files.
Classifier Data¶
SpliceGrapher creates three files for each classifier: a .svm file that contains
the trained classifier parameters; a .fa file that contains the sequences used
for training, and a .cfg file that contains meta-data about the classifier including
the parameters optimized by select_model_parameters.py and its ROC score.
To avoid having to track three files for every classifier, SpliceGrapher usually
stores these files in a single convenient .zip archive.
Generating ROC Curves¶

Example of an ROC curve generated by generate_roc.py for a GT classifier.
SpliceGrapher includes a script for generating ROC curves for classifiers.
The basic command syntax is generate_roc.py config-file. This script reads
the files generated by the parameter selection program, performs 5-fold cross-
validation, and generates an ROC curve. For example:
generate_roc.py gt_don.cfg -o gt_roc.pdf
Example output is shown in the figure. It may not always be possible
to train a classifier with strong ROC scores. In these cases it may be useful to
set a decision function threshold for classifying splice sites. The generate_roc.py
script can aid in this process. Using the -D option, one
can see the True Positive rate associated with different decision function
values. One can then set an appropriate threshold value in the classifier
configuration (.cfg) file for classifying splice sites.
Predicting Splice Graphs¶
Splice graph prediction is SpliceGrapher’s main purpose. This process involves two key aspects: validating the available evidence and making confident predictions from all available evidence.
Filtering Spliced Alignments¶
A key aspect of splice graph prediction is ensuring the quality of the evidence being used.
To this end, SpliceGrapher includes scripts for filtering spliced alignments in the SAM
format. The first is sam_filter.py (which replaces filter_sam_jct.py in older versions).
It takes as input a SAM file and a set of classifiers and produces a copy of the SAM file
with false-positive sites removed:
sam_filter.py alignments.sam classifiers.zip -o filtered.sam
The script accepts the following options:
| Option | Value | Description |
|---|---|---|
| -F | SAM file | File for storing false-positive records |
| -o | OUTPUT | Output file |
| -r | REPORT | Write classifier scores to file |
| -z | Apply gzip compression to output |
By default, SAM records that include false-positive sites will be discarded. You
may save these records to a separate file using the -F option.
The -r option allows you to see the classifier scores for each splice site
found in the input file: negative values show which sites were predicted as
false-positives.
Collating SAM Files¶
For large volumes of RNA-Seq data, a single SAM file may be unwieldy. For this
reason SpliceGrapher includes the script sam_collate.py that reads a SAM
alignment file and writes the output to separate files, one for each chromosome.
sam_collate.py *.sam
Because SAM files can be large, the script can read gzipped files and with the
-z option, it can write them as well.
Note: Most SpliceGrapher scripts expect SAM files to be sorted by chromosome and position within the chromosome. A command for sorting a SAM file is:
sort -k 3,3 -k 4,4n unsorted.sam > sorted.sam
Alternately you may use samtools for manipulating files prior to collating.
SpliceGrapher Depths Files¶
With version 0.2.5 we introduced SpliceGrapher depths files.
These files encapsulate the coverage information from a SAM or BAM file in a
compact format that SpliceGrapher can then use to make predictions or to generate plots.
These depths files may be up to two orders of magnitude smaller than the original SAM or BAM
file (e.g., from 5GB to 50MB) and thus can dramatically reduce
running times for other SpliceGrapher scripts.
To create a depths file from a sorted SAM or BAM file,
use the sam_to_depths.py script:
sam_to_depths.py accepted_hits.bam -o accepted_hits.depths
Once a depths file has been created, you may use it in place of SAM files in other SpliceGrapher scripts, for example:
predict_graphs.py accepted_hits.depths -m h_sapiens.gtf -d predicted
Similarly, you may use a depths file in a plotter.py configuration file:
[Reads]
plot_type = read_depth
source_file = accepted_hits.depths
title_string = %gene Read Coverage
Thus, sam_to_depths.py creates compact files that allow other scripts to run
faster, and permits you to use SpliceGrapher equally well with either BAM or SAM files.
Note: to use sam_to_depths.py you must install the pysam package,
available from code.google.com/p/pysam.
Creating Splice Graphs from Gene Models¶
Splice graphs are a key data structure for the SpliceGrapher package. They can be used to depict a gene model, a collection of EST alignments and of course, they provide the foundation for splice graph predictions. SpliceGrapher files are stored in a GFF format that is specific to these graphs.
The script gene_model_to_splicegraph.py accepts a GFF3 gene model annotation
file as input and generates splice graphs as output. The output file may contain
all the genes in the gene model file, or you may specify an output directory and
write a separate file for each gene. For example, to generate splice graphs for
all gene models in the GENE_MODEL file (from SpliceGrapher.cfg), the
command would be:
gene_model_to_splicegraph.py -o gene_models.gff
To generate the same graphs into separate files in a directory
/home/mygraphs, the command would be:
gene_model_to_splicegraph.py -d /home/mygraphs
It is often useful to organize splice graphs by chromosome, since reference
sequences are separated that way. For example, instead of having a single top-
level directory, you may have a directory for each chromosome, such as
/home/mygraphs/chr1, /home/mygraphs/chr2, .... In this case, simply use the
-c option to specify the chromosome for which you want to generate graphs:
gene_model_to_splicegraph.py -d /home/mygraphs/chr3 -c chr3
Finally, you may use the script to generate splice graphs for a specific set of
genes using the -g option. For example, if you were interested in generating
graphs only for the Arabidopsis genes AT3G01100, AT3G01120, AT3G01460 and
AT3G01490 into the local directory you would enter:
gene_model_to_splicegraph.py -d . -g AT3G01100,AT3G01120,AT3G01460,AT3G01490
Note: although GFF3 gene model annotations are becoming more standardized, not all organisms adhere to the same naming conventions. For example, UTR records may be given as ‘three_prime_UTR’ and ‘five_prime_UTR’, or simply ‘UTR’ in which case the relative gene position must be inferred. Thus you may need to modify the GFF3 annotations prior to converting them to splice graphs if you want access to all gene features. (See the GeneModel module for more information.)
Converting GTF Annotations¶
GTF file functionality allows users to work
easily with gene models in either GTF or GFF3 formats.
We have replaced the gtf2gff.py script with convert_models.py
that can convert gene models in GTF format to GFF3 format and vice versa.
To convert from GTF to GFF3, the command is:
convert_models.py Homo_sapiens.gtf --gff3=Homo_sapiens.gff3
To convert from GFF3 to GTF, the command is:
convert_models.py Homo_sapiens.gff3 --gtf=Homo_sapiens.gtf
Note: for both GTF and GFF3 files, SpliceGrapher expects curated gene models from sites such as UCSC, ENSEMBL or the TAIR website. In many cases it can accept GTF files output from tools like Cufflinks, but this behavior is not guaranteed, as not all GTF or GFF3 files represent gene models. Also, some tools may not adhere strictly to the GTF or GFF3 standards. For more on these file formats, see GTF.
Creating Splice Graphs from ESTs¶
Popular EST alignment algorithms such as BLAST, BLAT and GMAP can produce output
in PSL format. Consequently SpliceGrapher includes a script,
ests_to_splicegraph.py that converts EST alignments in PSL format to a splice
graph. The script operates very much like the gene_model_to_splicegraph.py
script outlined above except that you must specify a PSL file:
ests_to_splicegraph.py gmap_alignments.psl -d est_splicegraphs
As before, you may specify a single output file or an output directory, and you may request specific gene names.
Alignment programs sometimes yield introns that are too short (for example exons may abut one another) and that may lead to spurious alternative splicing predictions. To address this issue, the script allows you to specify a minimum allowed intron length (default is 4nt). For example, if the smallest known intron for an organism is 10nt long, you might use:
ests_to_splicegraph.py gmap_alignments.psl -d est_splicegraphs -i 10
Alignments in the PSL file that infer introns shorter than this will not be included in the splice graphs.
predict_graphs.py¶
SpliceGrapher allows you to predict the splice graphs
for every gene in a file of gene models at once.
The predict_graphs.py script provides a simple interface for loading
RNA-Seq alignments along with gene models to generate predictions for all
genes that have read coverage.
The predicted graphs will be created in files stored in subdirectories
named for each chromosome in the gene models.
The basic format of the command is:
predict_graphs.py alignments-file [options]
Here, alignments-file may either be a SAM file, or starting with version 0.2.5
you may use a SpliceGrapher depths file (see SpliceGrapher Depths Files).
This program accepts the following parameters:
| Option | Value | Description |
|---|---|---|
| -d | Output dir | Top-level directory to hold output graphs |
| -m | Gene models | short-read alignments |
| -J | JMINDEPTH | Minimum coverage required across splice junctions |
| -T | THRESHOLD | Minimum depth threshold for accepting short-read clusters |
--mindepth |
MINDEPTH | Average exon depth required to make a prediction |
With version 0.2.7, users may now use the --mindepth option to specify a minimum
exon depth that will be required before SpliceGrapher predicts any AS for a gene.
This helps to reduce potentially spurious predictions that may arise in genes with
weak or highly variable read coverage.
Note: when used with SAM files, predict_graphs.py loads the complete
file along with a complete set of gene models, so it can require a lot of memory
(40GB or more for predictions across the human genome). To avoid this issue,
first convert the SAM file into a SpliceGrapher depths file (SpliceGrapher Depths Files).
Alternately you may split the SAM file (see Collating SAM Files) and gene models
by chromosome and run predict_graphs.py on each chromosome separately.
predict_splicegraph.py¶
To predict splice graphs from diverse evidence sources, SpliceGrapher
uses the script predict_splicegraph.py.
Given annotations for a particular gene along with
additional evidence such as EST alignments,
ungapped short read alignments and splice junction alignments,
it combines the information and uses inference rules to predict an output graph.
The program starts with a splice graph created from a gene model. To this you may add additional splice graphs (such as those generated from EST alignments) or short read alignment data. The output is a single splice graph that incorporates all of the data sources into a single predicted graph.
The basic format of the command is:
predict_splicegraph.py gene-model [options]
This program accepts the following parameters:
| Option | Value | Description |
|---|---|---|
| -d | SAM file | short-read alignments |
| -J | JMINDEPTH | Minimum coverage required across splice junctions |
| -M | MINANCHOR | Minimum anchor size required for junction evidence |
| -s | GRAPHS | Comma-separated list of EST splice graph files |
| -T | THRESHOLD | Minimum depth threshold for accepting short-read clusters |
Use the -s option to include additional splice graphs from EST alignments. For example,
for the Human gene HES4, you might use:
predict_splicegraph.py HES4_model.gff -s HES4_ests.gff
To include RNA-Seq reads, specify a SAM alignment file using the -d option:
predict_splicegraph.py HES4_graph.gff -d chr1.sam.gz
The -M option provides flexibility beyond the anchor constraints placed on spliced
alignments. Even if you specify a minimum anchor size for spliced alignment, the
distribution of reads across a junction may not be uniform.
This option tells the prediction program to use only junctions supported by at least one read
with this many bases on each side:
predict_splicegraph.py HES4_graph.gff -d chr1.sam.gz -M 12
Finally, the -T option provides the prediction program with a threshold for treating
short-read clusters as background noise. By default, SpliceGrapher accepts all short-read
clusters as possible evidence.
Finding Splice Forms¶
SpliceGrapher also has the ability to identify annotated splice forms
that are represented in a set of RNA-Seq data.
It uses existing gene models to establish a set of known transcripts that
are comprised of lists of nodes from a splice graph.
By identifying splice junctions and nodes unique to each transcript, it can
infer which transcripts are present in a set of aligned reads.
The find_splice_forms.py script accepts as input a SAM file of RNA-Seq
alignments and reports splice forms from annotated gene models where the
RNA-Seq data provide sufficient evidence for them.:
find_splice_forms.py SAM-file [options]
The script uses the following parameters to assess all genes in a gene model file:
| Option | Value | Description |
|---|---|---|
| -d | OUTDIR | Output directory (overrides -o) |
| -j | MINJCT | Minimum junction threshold |
| -o | OUTPUT | Output file |
| -O | OVERLAP | Minimum number of bases that a read cluster must overlap a feature |
| -t | THRESHOLD | Minimum average read coverage for clusters |
Note that a gene model is absolutely required for making these predictions.
The input gene model file may either be given as a command-line option (-m)
or specified in your SpliceGrapher.cfg file.
Each splice form is represented by splice junctions or exons unique to that form.
find_splice_forms.py loads RNA-Seq alignments and converts them into clusters
of overlapping reads. When a cluster overlaps sequence that is unique to a
single exon and that exon is unique to a single splice form, the splice form will
be included in the final graph. Similarly, when spliced reads fall across a
splice junction that is unique to a particular splice form, the splice form will
be included in the final graph.
Three parameters control the sensitivity/specificity of this program: minimum
average cluster read coverage (-t), a minimum number of splice junction reads (-j),
and the minimum required cluster overlap (-O).
Read clusters must have the desired minimum coverage before they are used to identify
splice forms.
Likewise, splice junctions must have the given minimum number of reads before they
are used to identify forms.
In cases where the RNA-Seq coverage does not uniquely cover any particular splice
forms, no output graph will be produced.
You may specify an output file (-o), in which case all splice graphs will be written to
the same file.
The recommended approach is instead to specify an output directory (-d) in which case
a separate file will be written for each gene for which a graph is produced.
Features That Uniquely Identify Splice Forms
In this context, a feature is that part of an exon that is completely unique to a particular splice form. Clusters must overlap unique exon features by a minimum amount (the default is 1, but a higher number is recommended for better specificity). For example, an exon that represents a retained intron will typically have as its unique feature that region from the start to the end of the associated intron. Some unique features may be smaller than the minimum overlap, for example, when the minimum is set to 10 and an alternative 3’ site is a NAGNAG site whose unique feature is just 3nt long. For this reason, SpliceGrapher uses the smaller of the overlap size or the feature length.
Viewing Splice Graphs¶
Simple Viewing Tool¶
SpliceGrapher includes two scripts for viewing splice graphs. These scripts also
serve as examples of how to use the SpliceGrapher viewing modules, many of which
have been encapsulated in the ViewerUtils module.
The simplest way to view
one or more splice graphs is to use the view_splicegraphs.py script. For
example, to view the graph stored in gene_graph.gff, simply enter:
view_splicegraphs.py gene_graph.gff
This produces a splice graph with minimal annotations. The script will
accept more than one file on the command line. For example, to plot the files
AT1G01020.gff, AT1G01030.gff, AT1G01040.gff and AT1G01060.gff, one might
enter either of the following commands:
view_splicegraphs.py AT1G010[2346]0.gff
view_splicegraphs.py AT1G010*.gff
This can be powerful if you want to compare graph predictions for the same gene across different data sets (such as for different tissue samples or mutants):
view_splicegraphs.py data/var[1-3]/AT1G01020.gff
The script provides a selection of options that allow you to make the output graph as elaborate as you like:
| Option | Value | Description |
|---|---|---|
| -a | (Re)annotate graphs | |
| -m | MODEL | GFF gene model reference |
| -o | OUTPUT | Output file |
| -x | Show genomic positions of graph elements | |
| -E | EDGE | Intron edge weight |
| -H | HEIGHT | Display area height, in inches |
| -W | WIDTH | Display area width, in inches |
| -L | Add legend to splice graph plot | |
| -F | FONTSIZE | Font size for plot titles |
| -X | Use the same genomic position range for each plot | |
| -S | Shrink introns |
For example, to generate a more elaborate plot for the comparison above and write the output to a PDF file, one could enter:
view_splicegraphs.py data/var[1-3]/AT1G01020.gff -xL -t var1,var2,var3 -o comparison.pdf
Shrinking Introns¶
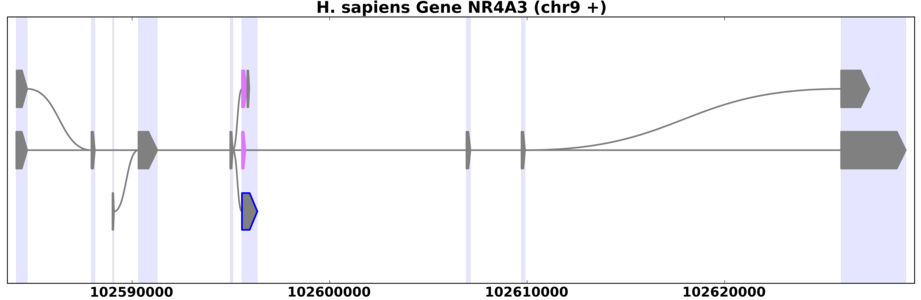
Example of a splice graph for a human gene with large introns.
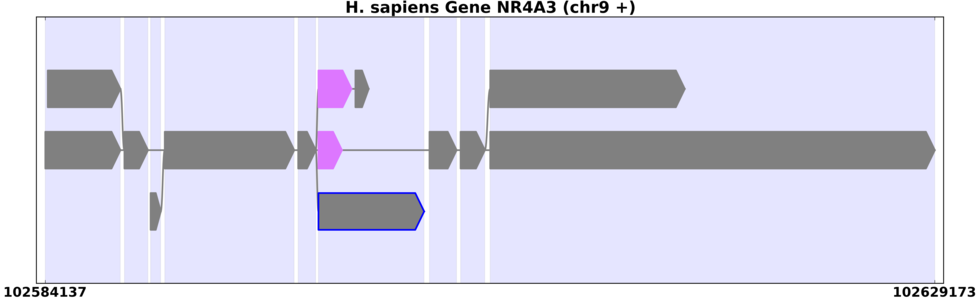
The same human gene with introns reduced to emphasize exons.
Many organisms, especially mammals, have introns that are much longer than exons. This can make it difficult to see alternative splicing behavior on these plots. Figure 8.1 shows an example of a human gene that has large introns relative to its exons as generated from the command:
view_splicegraphs.py -m chr9.gff.gz NR4A3
You can change the way these appear in view_splicegraphs.py using the -S option (Figure 8.2):
view_splicegraphs.py -m chr9.gff.gz NR4A3 -S
Plot Types¶
SpliceGrapher can produce different kinds of plots for different kinds of data. These are: splice graphs, isoform plots, read coverage plots, splice junction plots and X-Y plots. These are described in more detail below:
Splice Graphs¶
Splice graphs are SpliceGrapher’s principal plot type. These depict splice graphs that represent all the different ways in which a gene’s exons may be combined.
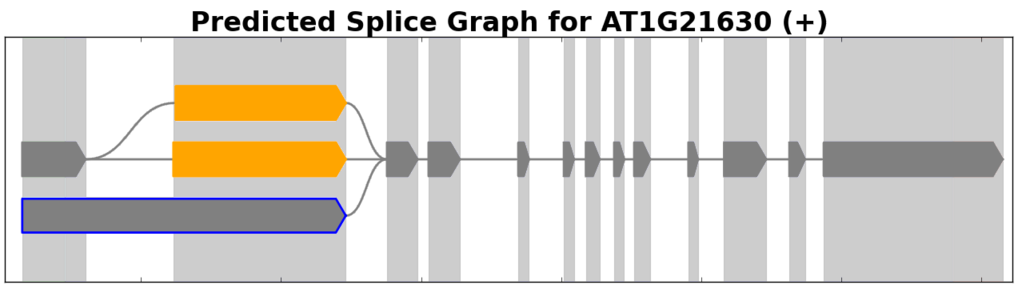
Example of a splice graph for a gene from the plant A. thaliana.
Isoforms¶
Isoform graphs are similar to splice graphs in the way that introns and exons are depicted. However, instead of plotting the graph in its compact representation, isoform graphs show each possible splice form from a graph. These are particularly useful for showing splice forms represented in RNA-Seq data (see Finding Splice Forms).
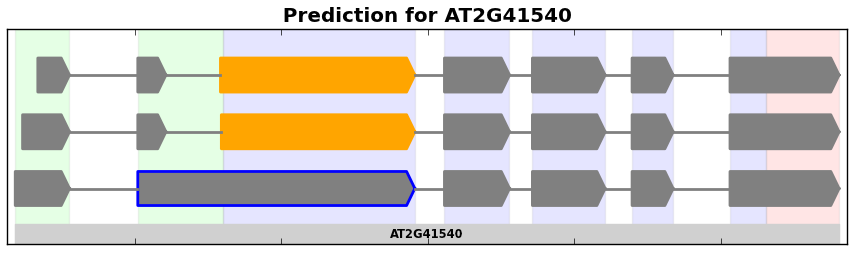
Example of an isoform graph for a gene from the plant A. thaliana.
Read Coverage¶
Read coverage graphs depict the way RNA-Seq reads aligned to a gene region. The height of the graph at any given position represents the number of reads whose alignments overlap at that point.
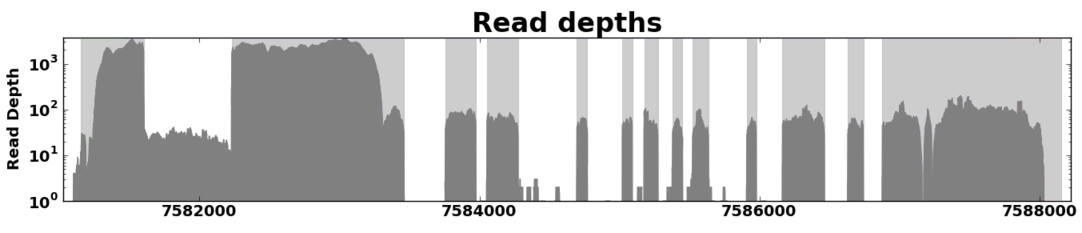
Example of a read coverage graph for a gene from A. thaliana.
Splice Junction Coverage¶
Splice junction coverage graphs depict the way RNA-Seq reads aligned across splice junctions within a gene region. Splice junctions are depicted as ‘^’-shaped widgets flanked by bars that depict the anchor regions on either side of a junction. Labels for each splice junction show the number of reads that aligned across each junction.
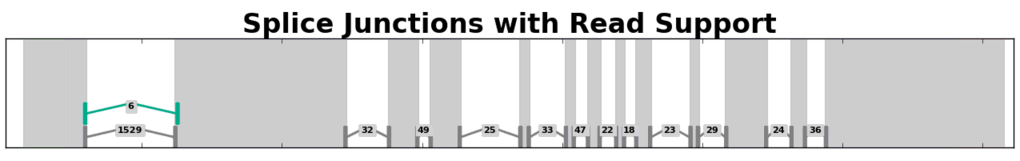
Example of a splice junction coverage graph for a gene from A. thaliana.
X-Y Plots¶
SpliceGrapher also provides a generic X-Y plot that can depict arbitrary values across a gene region. These could be used, for example, to plot values from other forms of genomic activity (such as methylation) to see if there is any correlation between alternative splicing and these other phenomena.
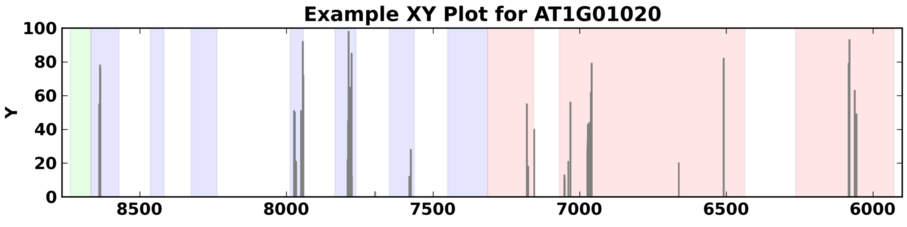
Example of an X-Y graph for a gene from A. thaliana using randomly-generated values between 1 and 100 to simulate activity within the gene.
Multiple Plots¶
For more elaborate plots, SpliceGrapher includes a second script designed to show a predicted splice graph along with its accompanying evidence. It accepts a variety of data formats as input and can produce pages with one to four plots. Each of these plots requires a separate input file.
- Predicted splice graph (SpliceGrapher GFF format)
- Original gene model splice graph (SpliceGrapher GFF format)
- Short read depths (SAM format)
- (Optional) splice junctions with short read support (SAM format)
File Options¶
The options for this script are organized into file options and display options. File options tell the script what the kinds of input files to expect and how to render the output graphs. You may include any combination of input files and display them in any order (see Display Options below).
| File Options | Value | Description |
|---|---|---|
| -m | MODEL | Gene models (GTF or GFF3 format) |
| -o | OUTPUT | Output plot file (infers format from file extension) |
| -d | DEPTH_FILE | SAM alignments (ungapped and spliced reads) |
| -s | SPLICE_GRAPH | GFF splice graph file |
| -G | ORIG_GRAPH | Optional original graph file |
| -X | XYDATA | Data file of X,Y value pairs for a simple graph |
Note: SAM alignments may be specified in two ways. Some pipelines may produce separate files for ungapped alignments and spliced alignments, in which case both the -d and -j options should be used. Other methods (notably TopHat) produce a single file that contains both ungapped and spliced alignments, in which case just the -d option should be used.
Display Options¶
The display options are nearly the same as those for view_splicegraphs.py:
| Display Options | Value | Description |
|---|---|---|
| -c | Add read coverage labels to junctions | |
| -x | Add genomic position labels to plots | |
| -E | EDGE | Minimum edge thickness |
| -J | MINJCT | Minimum junction depth |
| -L | Add a legend to graph | |
| -H | HEIGHT | Display window height (inches) |
| -W | WIDTH | Display window width (inches) |
| -F | FONTSIZE | Font size for plot titles |
| -S | Shrink introns relative to exons | |
| -D | DISPLAY | Display order string for plots |
As an example, suppose we have used SpliceGrapher to predict a splice graph
for the gene G123. We used gene_model_to_splicegraph.py to generate a splice
graph for the gene model and stored it in G123m.gff, generated splice
junction and read depth files in SAM format and used predict_splicegraph.py
to generate a splice graph prediction and stored it in G123p.gff. To produce
a four-panel plot of the predicted graph along with its evidence, we could
enter:
view_splicegraph_multiplot.py G123 -d chr1.sam -G G123m.gff -s G123p.gff
Note that the background for each of the panels includes a color-coded synopsis
of the gene model. Thus we may choose to omit the gene model graph from the
output and focus instead on the prediction and its evidence. In addition,
suppose we wish to see the evidence at the top of the page and the predicted
splice graph at the bottom. We may change all of these characteristics in a
single step using the -D option. It accepts a string that determines the
display order and whether or not to display each plot. Each character represents
a specific plot: ‘O’ for the original gene model, ‘P’ for the predicted splice
graph, ‘J’ for the splice junction plot, and ‘R’ for the read depth plot. The
default display order is ‘OPJR’, so to modify a graph to omit the original graph
and change the order, simply enter the string ‘RJP’:
view_splicegraph_multiplot.py G123 -d chr1.sam -G G123m.gff -s G123p.gff -D RJP
High-Quality Plotting¶
In addition to viewing utilities, SpliceGrapher provides plotting utilities
that allow you to tune your plotting parameters for a specific purpose (such
as figures suitable for a talk or paper) and save your configuration for later use.
The program is called simply plotter.py and has the following format:
plotter.py gene.cfg
The plotting utility has a few options that allow you to try different output parameters such as page dimensions, but because the program provides a rich set of options, it is more convenient to store these in a configuration file. Here is an example:
[SinglePlotConfig]
legend = True
shrink_introns = True
height = 5.0
width = 18.0
fontsize = 16
output_file = NR4A3.pdf
[GeneModelGraph]
plot_type = gene
gene_name = NR4A3
relative_size = 10.0
source_file = ${GENE_MODELS}/chr9.gff3.gz
file_format = gene_model
title_string = Gene Model for %gene
[SpliceGrapher]
plot_type = splice_graph
relative_size = 10.0
source_file = ${GRAPHS}/NR4A3_pred.gff
title_string = SpliceGrapher Prediction for %gene
[Reads]
plot_type = read_depth
relative_size = 8.0
source_file = ${SAM}/chr9.sam.gz
title_string = %gene Read Coverage
Main Section¶
The only required section is the main [SinglePlotConfig] section that provides
general parameters for producing a single plot of one or more panels. The
remaining sections each identify a distinct panel that will appear on the same plot in the
output file. You may specify as many of these panels as you wish, provided each one has
a unique name. The main difference between the graph sections and the main section is the
parameters that are allowed for each section. SpliceGrapher enforces strict typing on these
parameters and values.
Parameters for the [SinglePlotConfig] section are:
| Name | Type | Description |
|---|---|---|
| fontsize | int | Font size for title strings |
| legend | boolean | Include legend on all plots |
| log_file | string | Write error messages to the given log file instead of stderr |
| height | float | Page height (in inches) |
| output_file | string | Path to output file |
| shrink_introns | boolean | Shrink introns relative to exons |
| width | float | Page width (in inches) |
Plot Sections¶
Parameters for individual graphs are:
| Name | Type | Description |
|---|---|---|
| acceptors | string | Comma-delimited list of acceptor sites to highlight (junctions only) |
| background | boolean | Show the gene model in the plot background |
| clusters | boolean | Show read depths as whole clusters (read_depth only) |
| donors | string | Comma-delimited list of donor sites to highlight (junctions only) |
| edge_weight | int | Edge weight to use when drawing intron edges |
| file_format | string | Type of data file to use (gene_plot only) |
| gene_name | string | Gene name to use (gene_plot only) |
| hide | boolean | Use the gene information but do not display the plot (gene_plot only) |
| highlight | string | Comma-delimited list of regions to highlight (read_depth only) |
| labels | boolean | Plot-dependent labeling: exon ids for splice graphs, read coverage for junctions |
| min_coverage | float | Minimum read coverage required for showing junctions |
| relative_size | float | Relative plot size |
| plot_type | string | One of: gene/isoforms/junctions/read_depth/splice_graph/xy_plot |
| source_file | string | Path to file containing data for this plot |
| title_string | string | Text to use for plot title |
| x_labels | boolean | Show positions for all features in plot |
Note: A gene model section is required for all plots that use
shaded bars in the background. To prevent the gene model itself from
plotting, use the hide option.
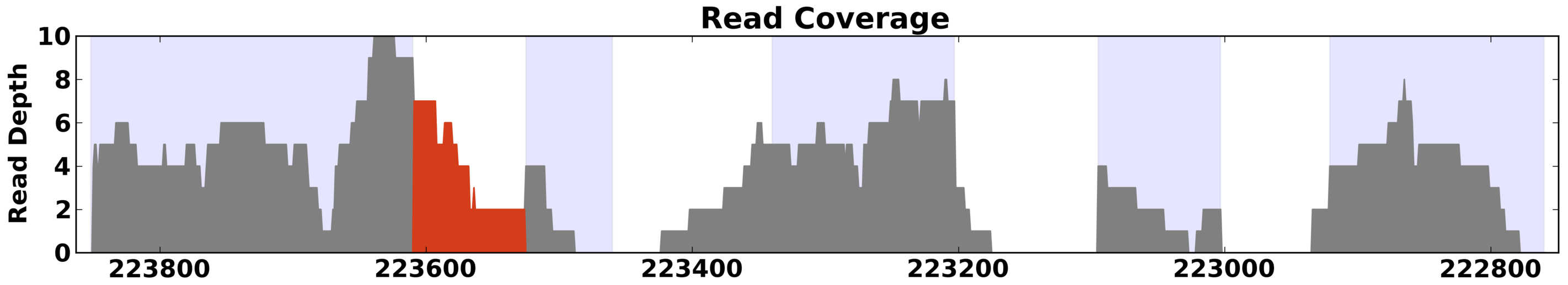
Example of read depth plot with highlighting turned on to highlight possible intron retention activity.
Source Files for Plots¶
Each plot type is associated with specific file formats:
| Plot Type | File Format | Description |
|---|---|---|
| gene | gene model GFF | Plots a gene model as a splice graph |
| isoforms | splice graph GFF | Plots elements in a splice graph as distinct splice forms |
| junctions | SAM/depths | Depicts all splice junctions from alignments |
| read_depth | SAM/depths | Depicts read coverage based on alignments |
| splice_graph | splice graph GFF | Plots a splice graph |
| xy_plot | CSV | Plots X-Y values |
Note: if you use SAM files, the plotting tools expect them to be sorted.
If you provide an unsorted SAM file to plotter.py,
you may see nothing in the splice
junctions or read depth sections of your plot. See Collating SAM Files for
information on how to collate and sort SAM files.
Output Options¶
Although the configuration file allows you to specify output settings such
as page width and height and font size, you may wish to try different settings
to get a figure to look the way you want. Thus plotter.py provides
command-line options to change these settings:
| Option | Value | Description |
|---|---|---|
| -F | FONTSIZE | Font size (12-point) |
| -H | HEIGHT | Plotting area width (11.0 inches) |
| -W | WIDTH | Plotting area width (8.5 inches) |
| -o | OUTPUT | Output file |
By default, a plot will appear on the screen, but if you specify an output
file, plotter.py will output to the file, using the file extension to
determine the file format.
Valid file extensions are: emf, eps, pdf, png, ps, raw, rgba, svg and svgz.
These command-line settings will override settings in the configuration file.
If no settings are specified in either place, the defaults are used.
Realignment Pipeline¶
Initially, SpliceGrapherXT predicts novel exons conservatively and records information about those it could not resolve due to ambiguous combinations of acceptor and donor sites. We wish to resolve those ambiguous loci by realigning RNA-Seq reads to a database of putative transcripts in order to find compelling evidence for exons that SpliceGrapherXT could not resolved in its inference step. We thus created a pipeline that constructs putative transcripts from unresolved exons, aligns reads to these transcripts, and resolves exons that have sufficient coverage from the alignments.
The pipeline proceeds in three stages: first it generates putative transcripts using unresolved exons; next it realigns reads to the putative transcripts using BWA, and finally it resolves novel exons when realigned reads completely span the exon and its boundaries.
The script for running the pipeline is realignment_pipeline.py
and has the following format:
realignment_pipeline.py original-dir
Here the original directory is the directory containing original
predictions created with predict_graphs.py. The realignment
pipeline may be used with either single-end or paired-end reads,
as follows. For single-end read, merely specify a single FASTQ file:
realignment_pipeline.py original_predictions -1 myreads.fq
For paired-end reads, specify two FASTQ files, where the second file contains the mate pairs for the reads in the first file:
realignment_pipeline.py original_predictions -1 myreads_1.fq -2 myreads_2.fq
The pipeline script has just a few options:
| Option | Value | Description |
|---|---|---|
| -1 | FASTQ | Reads file or first set of mate pairs |
| -2 | FASTQ | Second set of mate pairs (paired-end only) |
| -d | OUTDIR | Output directory for updated predictions |
| -f | FASTA | Genome reference file |
| -m | MODEL | Gene model annotations |
Below is output from an example run on 100-nt paired-end reads (some paths have been shortened for readability)
realignment_pipeline.py sgxt_pred -1 reads_1.fq -2 reads_2.fq -v -d realigned
03:46:33 realignment_pipeline.py Started
Generating putative transcript database
03:46:33 generate_putative_sequences.py sgxt_pred.lis -Uo putative.fa
-m putative_transcripts.map
Running BWA alignments on FASTQ files using 4/8 CPUs
03:46:35 bwa aln -f reads_1.aln -t 4 -M 8 -R 2 -o 0 -e 0 -d 0
-i 100 putative.fa reads_1.fq
03:46:38 bwa aln -f reads_2.aln -t 4 -M 8 -R 2 -o 0 -e 0 -d 0
-i 100 putative.fa reads_2.fq
03:46:42 bwa sampe -n 2 -N 2 -P -f pair_1.raw putative.fa
reads_1.aln reads_2.aln reads_1.fq reads_2.fq
03:46:49 get_good_pairs.py pair_1.raw pair_1.pairs
Updating original predictions.
03:47:10 fix_unresolved.py sgxt_pred.lis putative_transcripts.map
filtered_bwa.sam -f putative.fa -c -d realigned -v
Finished.
The pipeline script shows each of the commands it uses as it runs. Note that it expects the
BWA alignment software to be in the user’s path. The first step generates a putative transcript
database that consists of two files: putative.fa is a FASTA file that contains the putative
sequences along with metadata in the sequence headers such as unique identifiers that
aid in post-processing.
putative_transcripts.map provides a mapping between the transcript identifiers and the splice graph
nodes from which they were generated.
The second step is the BWA alignment phase, in which FASTQ reads are aligned to the putative transcriptome. In the case of paired-end reads, there is an additional step to ensure that read alignments adhere to strict constraints. The final step updates the graphs by searching for unresolved exons that may be resolved when the aligned reads completely cover an exon and its boundaries. The updated graphs are then placed in the output directory. See [ROGERS2013] for more details on the realignment procedure.
Transcript Prediction Pipelines¶
SpliceGrapherXT also provides two methods for predicting transcripts that are novel relative to the input gene models. The basis of these methods is to use predicted splice graphs to generate a set of putative transcripts that we can provide to one of two other methods—PSGInfer or IsoLasso—to predict transcript frequencies or expression levels. SpliceGrapherXT then predicts novel transcripts whenever a putative transcript has a substantial frequency or estimated expression level. The approach is similar to the realignment procedure, but instead of generating putative transcripts only for graphs with unresolved nodes, it generates transcripts by traversing all the paths in every graph that has multiple paths. In this case, these putative transcripts are stored as a set of gene models that are then provided to other tools to predict either probabilistic splice graphs or transcript expression levels.
In principle this approach could used with any tool that predicts transcript expression levels. SpliceGrapherXT provides pipelines for two such approaches: PSGInfer (see [LEGAULT2013]) and IsoLasso (see [LI2011]).
PSGInfer Pipeline¶
The PSGInfer pipeline uses a list of splice graphs and two FASTQ files as input, and produces splice graphs that include novel predicted transcript annotations:
psginfer_pipeline.py graph-list fastq1 fastq2
Example:
psginfer_pipeline.py initial.lis reads_1.fq reads_2.fq
The script accepts the following options:
| Option | Value | Description |
|---|---|---|
| -d | OUTDIR | Output directory for updated predictions |
| -f | FASTA | Genome reference file |
| -l | LOGFILE | Optional log file [default: none] |
| -t | THRESH | Probability threshold [default: 0.01] |
Below is output from an example run on 100-nt paired-end reads (some paths have been shortened for readability):
psginfer_pipeline.py sgxt_pred.lis reads_1.fq reads_2.fq -d psg_updated
-t 0.15 -v -l psginfer_update.log
05:45:41 psginfer_pipeline.py Started
05:45:51 generate_putative_sequences.py sgxt_pred.lis -M putative_forms.gtf
-m putative_forms.map -UA
Changing directories to /tmp/test/FASTA_REF
05:45:54 psg_prepare_reference.py putative_forms.gtf putative_forms_psg_reference
05:46:43 psg_infer_frequencies.py reads_1.fq reads_2.fq
putative_forms_psg_reference putative_forms_psg_results --num-CPUs=2
Changing directories back to /tmp/test
05:52:01 psginfer_update_graphs.py sgxt_pred.lis putative_forms.map
FASTA_REF/putative_forms_psg_results/isoform.results.txt
-vd psg_updated -t 0.15
Finished.
The first task in this pipeline is to generate a set of putative transcripts, including those that involve unresolved exons. As in the realignment pipeline, this creates a FASTA file of transcript sequences, along with a file that maps transcript identifiers to their corresponding splice graph nodes. Next it runs the PSGInfer scripts that prepare reference graphs and infer frequencies over those graphs from the RNA-Seq data (for details, see [LEGAULT2013]). In the last step, SpliceGrapherXT infers legitimate transcripts using the frequencies assigned by PSGInfer.
Note: the SpliceGrapherXT pipeline requires PSGInfer version 1.1.3.
IsoLasso Pipeline¶
The IsoLasso pipeline is similar to the PSGInfer pipeline, but uses alignments in SAM format instead of short reads:
isolasso_pipeline.py graph-files SAM-file
The pipeline provides access to two IsoLasso methods: the original method and a newer CEM algorithm. Both methods are highly dependent on their input parameters. The pipeline uses default parameters that have been tuned extensively on our own data; however, the pipeline includes a pass-through mechanism that allows users to access IsoLasso parameters if desired.
| Option | Value | Description |
|---|---|---|
| -C | Use CEM instead of LASSO | |
| -d | OUTDIR | Output directory |
| -p | PARAMS | Additional parameters for IsoLasso |
| -t | THRESH | Minimum FPKM threshold |
| -U | Include unresolved transcripts |
Example using the original IsoLasso method:
isolasso_pipeline.py sgxt_pred.lis filtered.sam -d psg_updated -t 100
Example using the IsoLasso/CEM method:
isolasso_pipeline.py sgxt_pred.lis filtered.dam -Cd cem_updated -t 100
Example output:
11:15:00 isolasso_pipeline.py Started
11:15:00 generate_putative_sequences.py sgxt_pred.lis -AU
-M initial_predictions_forms.gtf -m initial_predictions_forms.map
11:15:02 gtfToGenePred initial_predictions_forms.gtf stdout | genePredToBed |
sort -k1,1 -k2,2n > initial_predictions_forms.bed
11:15:03 runlasso.py -x initial_predictions_forms.bed --forceref --useem
-o cem_update filtered.sam
11:15:14 isolasso_update_graphs.py initial_predictions.lis initial_predictions_forms.map
cem_update.pred.gtf -t 1.00 -d cem_update
Finished.
Both the PSGInfer and IsoLasso pipelines will add new transcript identifiers to the splice graphs whenever the associated method predicts a sufficient expression level for a novel transcript (based on the threshold). These updated graphs may then be converted back into gene models and used, for example, to assess differential expression levels.
File Formats¶
Given the variety of data used to generate predictions, SpliceGrapher uses several different file formats to process data.
Gene Model Files¶
GFF3 Files¶
Gene models are often stored using the GFF3 format and may contain a wide variety of different record types, though SpliceGrapher uses only a subset of these types:
- chromosome records are used to establish boundaries for all genes within a chromosome, but may also be inferred from the genes found within a chromosome.
- gene records are required to establish parent/child relationships within a gene using the “ID” attribute in the last column. A gene record is assumed to have one or more children given as exon records.
- mRNA records are used to infer exon and intron boundaries as well as parent/child relationships. Each record must have an “ID” attribute and a “Parent” attribute that refers to its gene. An mRNA record is assumed to have one or more children given as UTR or CDS records.
- exon records are used to infer exon and intron boundaries as well as parent/child relationships. Each record must have an “ID” attribute and a “Parent” attribute that refers to its gene.
- CDS records (CDS, UTR, FIVE_PRIME_UTR, or THREE_PRIME_UTR) are used to infer exon and intron boundaries as well as parent/child relationships. Each record must have an “ID” attribute and a “Parent” attribute that refers to its mRNA parent.
GTF Files¶
The annotations for many organisms (notably those on the ENSEMBL website) use GTF format instead. SpliceGrapher can load either GFF3 or GTF files directly.
Splice Graph Files¶
SpliceGrapher also uses the GFF format to store splice graphs. The GFF format is a convenient way to express all the parent/child relationships in a graph, along with annotations for alternative splicing analysis and visualization purposes. A SpliceGrapher GFF file uses the following record types:
- graph records define a splice graph. These records include attributes that include the gene name and a brief description of how the splice graph differs from the gene models. Note that this replaces the old cluster name, which has been deprecated.
- parent records define exons in a splice graph that act as root nodes in the graph.
- child records define all other exons in a graph, including leaf nodes.
Both parent and child records include attributes that describe unique identifiers for each exon, the splicing forms that include them, and the alternative splicing forms associated with them. Child records also include their parents’ unique identifiers. Many of these attributes are used by SpliceGrapher’s visualization modules.
SAM and BAM Files¶
SAM files are similar to GFF files but are used to store short-read depths after
reads have been aligned to a genome (for a complete description, see
http://samtools.sourceforge.net/SAM1.pdf).
BAM files are simply compressed, binary versions of SAM files.
With version 0.2.5, SpliceGrapher provides the sam_to_depths.py script
for converting SAM or BAM files into an even more compact depths format
(see SpliceGrapher Depths Files).
SpliceGrapher uses short read
alignments stored in the SAM format or depths format
to predict splice graphs and to display read
depths and splice junctions on plots.
BED and WIG Files¶
Some spliced alignment tools use BED and WIG files to store information about short-read alignments and splice junctions that have short-read support. SpliceGrapher can read these files to predict splice graphs and to display read depths and splice junctions on plots. For more information on these file formats, see http://genome.ucsc.edu/FAQ/FAQformat.html.
PSL Files¶
Programs that perform EST or cDNA alignments, such as BLAST and GMAP, can produce alignment information in PSL format. SpliceGrapher uses PSL files to construct splice graphs from these alignments that may then be merged with other graphs to make predictions. For details on the PSL file format, see http://fungi.ensembl.org/info/website/upload/psl.html.
Pre-Built Classifiers¶
SpliceGrapher comes with a set of pre-built classifiers for over 100 different species. Only classifiers with adequate performance are included, so some species will have classifiers for canonical (GT donors/AG acceptors) and semi-canonical (GC donors) sites while others will have classifiers only for canonical sites. This table below shows the ROC scores corresponding to the each of the pre-built classifiers:
| Species | GT | GC | AG |
|---|---|---|---|
| Acyrthosiphon pisum | 0.96 | 0.94 | |
| Aedes aegypti | 0.96 | 0.95 | |
| Amphimedon queenslandica | 0.99 | 0.99 | |
| Anolis carolinensis | 0.98 | 0.96 | |
| Anopheles gambiae | 0.96 | 0.95 | |
| Apis mellifera | 0.98 | 0.96 | |
| Arabidopsis thaliana | 0.97 | 0.95 | |
| Ashbya gossypii | 0.99 | 0.96 | |
| Aspergillus clavatus | 0.98 | 0.93 | |
| Aspergillus flavus | 0.96 | 0.92 | |
| Aspergillus fumigatus | 0.97 | 0.90 | |
| Aspergillus fumigatusa1163 | 0.95 | 0.92 | |
| Aspergillus nidulans | 0.96 | 0.91 | |
| Aspergillus terreus | 0.97 | 0.93 | |
| Bombyx mori | 0.98 | 0.98 | |
| Bos taurus | 0.98 | 0.96 | |
| Botryotinia fuckeliana | 0.98 | 0.95 | |
| Brachypodium distachyon | 0.98 | 0.96 | |
| Caenorhabditis brenneri | 0.97 | 0.98 | |
| Caenorhabditis briggsae | 0.95 | 0.97 | |
| Caenorhabditis elegans | 0.97 | 0.98 | |
| Caenorhabditis remanei | 0.97 | 0.98 | |
| Canis familiaris | 0.98 | 0.95 | |
| Cavia porcellus | 0.97 | 0.96 | |
| Choloepus hoffmanni | 0.98 | 0.96 | |
| Ciona intestinalis | 0.98 | 0.96 | |
| Culex quinquefasciatus | 0.96 | 0.97 | |
| Danaus plexippus | 0.97 | 0.96 | |
| Danio rerio | 0.98 | 0.96 | |
| Daphnia pulex | 0.98 | 0.95 | |
| Dasypus novemcinctus | 0.97 | 0.96 | |
| Dictyostelium discoideum | 0.99 | 0.99 | |
| Dipodomys ordii | 0.98 | 0.97 | |
| Drosophila ananassae | 0.98 | 0.96 | |
| Drosophila erecta | 0.98 | 0.97 | |
| Drosophila grimshawi | 0.98 | 0.96 | |
| Drosophila melanogaster | 0.98 | 0.96 | |
| Drosophila mojavensis | 0.97 | 0.96 | |
| Drosophila persimilis | 0.96 | 0.96 | |
| Drosophila pseudoobscura | 0.98 | 0.96 | |
| Drosophila sechellia | 0.97 | 0.96 | |
| Drosophila simulans | 0.97 | 0.96 | |
| Drosophila virilis | 0.98 | 0.97 | |
| Drosophila willistoni | 0.97 | 0.96 | |
| Drosophila yakuba | 0.98 | 0.97 | |
| Echinops telfairi | 0.98 | 0.96 | |
| Entamoeba histolytica | 0.99 | 0.96 | |
| Equus caballus | 0.95 | 0.95 | |
| Erinaceus europaeus | 0.98 | 0.96 | |
| Felis catus | 0.98 | 0.96 | |
| Fusarium oxysporum | 0.96 | 0.92 | |
| Gadus morhua | 0.98 | 0.96 | |
| Gallus gallus | 0.97 | 0.95 | |
| Gasterosteus aculeatus | 0.96 | 0.95 | |
| Gibberella moniliformis | 0.97 | 0.93 | |
| Gibberella zeae | 0.98 | 0.94 | |
| Gorilla gorilla | 0.97 | 0.95 | |
| Heliconius melpomene | 0.96 | 0.96 | |
| Homo sapiens | 0.98 | 0.96 | |
| Ixodes scapularis | 0.94 | 0.93 | |
| Latimeria chalumnae | 0.97 | 0.94 | |
| Loxodonta africana | 0.96 | 0.95 | |
| Macaca mulatta | 0.95 | 0.93 | |
| Macropus eugenii | 0.97 | 0.96 | |
| Magnaporthe oryzae | 0.97 | 0.93 | |
| Meleagris gallopavo | 0.97 | 0.94 | |
| Microcebus murinus | 0.97 | 0.96 | |
| Mus musculus | 0.98 | 0.96 | |
| Myotis lucifugus | 0.97 | 0.97 | |
| Nematostella vectensis | 0.97 | 0.96 | |
| Neosartorya fischeri | 0.98 | 0.93 | |
| Neurospora crassa | 0.96 | 0.94 | |
| Nomascus leucogenys | 0.96 | 0.94 | |
| Ochotona princeps | 0.98 | 0.96 | |
| Ornithorhynchus anatinus | 0.96 | 0.94 | |
| Oryctolagus cuniculus | 0.97 | 0.96 | |
| Oryza sativa | 0.95 | 0.94 | |
| Oryzias latipes | 0.96 | 0.95 | |
| Otolemur garnettii | 0.97 | 0.96 | |
| Pan troglodytes | 0.98 | 0.96 | |
| Petromyzon marinus | 0.95 | 0.93 | |
| Physcomitrella patens | 0.98 | 0.95 | |
| Plasmodium berghei | 0.94 | 0.99 | |
| Plasmodium chabaudi | 0.96 | 0.98 | |
| Plasmodium falciparum | 0.96 | 0.98 | |
| Pristionchus pacificus | 0.94 | 0.98 | |
| Procavia capensis | 0.98 | 0.96 | |
| Pteropus vampyrus | 0.98 | 0.97 | |
| Pythium ultimum | 0.95 | 0.95 | |
| Rattus norvegicus | 0.95 | 0.93 | |
| Saccharomyces cerevisiae | 0.99 | 0.92 | |
| Sarcophilus harrisii | 0.97 | 0.95 | |
| Schistosoma mansoni | 0.96 | 0.94 | |
| Schizosaccharomyces pombe | 0.99 | 0.95 | |
| Sclerotinia sclerotiorum | 0.95 | 0.90 | |
| Sorex araneus | 0.98 | 0.96 | |
| Sorghum bicolor | 0.97 | 0.96 | |
| Sus scrofa | 0.97 | 0.96 | |
| Takifugu rubripes | 0.96 | 0.92 | |
| Tarsius syrichta | 0.99 | 0.97 | |
| Tetraodon nigroviridis | 0.96 | 0.95 | |
| Toxoplasma gondii | 0.98 | 0.98 | |
| Tribolium castaneum | 0.96 | 0.96 | |
| Trichoplax adhaerens | 0.96 | 0.93 | |
| Tupaia belangeri | 0.97 | 0.96 | |
| Tursiops truncatus | 0.98 | 0.97 | |
| Vicugna pacos | 0.98 | 0.96 | |
| Xenopus tropicalis | 0.96 | 0.94 | |
| Zea mays | 0.96 | 0.94 |
| [BENHUR2008] | (1, 2) Support vector machines and kernels for computational biology, A. Ben-Hur, C.S. Ong, S. Sonnenburg, B. Schölkopf and G. Rätsch, PLoS Computational Biology, vol. 4, no. 10, 2008. |
| [ROGERS2012] | SpliceGrapher: Detecting patterns of alternative splicing from RNA-seq data in the context of gene models and EST data, M.F. Rogers, J. Thomas, A.S.N. Reddy, and A. Ben-Hur, Genome Biology, 13(R4), 2012. |
| [ROGERS2013] | SpliceGrapherXT: from splice graphs to transcripts using RNA-Seq, M.F. Rogers, C. Boucher and A. Ben-Hur, ACM Conference on Bioinformatics, Computational Biology and Biomedical Informatics (ACM-BCB) 2013. |
| [LI2011] | IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly, Wei Li, Jianxing Feng and Tao Jiang, Journal of Computational Biology vol. 18, no. 11, 2011. |
| [LEGAULT2013] | (1, 2) Inference of alternative splicing from RNA-Seq data with probabilistic splice graphs. Laura H. LeGault and Colin N. Dewey, Bioinformatics. 2013. |